Sindrom Noonan
Apa arti noonan syndrome? Arti noonan syndrome atau NS pertama kali dijelaskan pada tahun 1963, ini adalah penyakit dominan autosomal (mempengaruhi kedua jenis kelamin secara setara) yang dapat ditularkan dari orang tua yang terkena ke beberapa anak mereka. Ekspresi tanda sangat bervariasi, tetapi subjek dengan sindrom noonan hampir selalu memiliki elemen sindrom yang ada (penetrasi hampir sempurna).
Fungsi pembelahan sel pada semua organisme multisel yaitu untuk pertumbuhan dan pemeliharaan serta perbaikan sel dan jaringan. Di NS, jelas bahwa fungsi pembelahan sel ini terganggu. Catatan, sindrom Noonan sebenarnya tidak dianggap penyakit langka, karena prevalensi sindrom ini saat lahir diperkirakan 1 dari 1000 sampai 1 dari 2500.
Etiologi noonan syndrome
Secara klasik ditandai dengan perawakan pendek, beberapa ciri hipertelorisme, morfologi minor dan kelainan bawaan (terutama jantung). Ini karena disfungsi sekelompok protein penting yang terlibat dalam pengendalian perkembangan, transmisi sinaptik dan respons seluler terhadap hormon pertumbuhan. Protein kunci untuk kontrol ini adalah protein RAS, itulah sebabnya sindrom noonan dan beberapa sindrom langka lainnya yang terkait dengan yang terakhir telah dikelompokkan bersama dibawah istilah “RASopathies”. Hipertelorisme adalah istilah yang digunakan untuk menggambarkan jarak yang sangat jauh di antara organ. Pada mata, ini mengacu pada posisi orbit tulang rongga mata, tempat mata terletak di tengkorak. Penglihatan hipertelorisme adalah sesuai dengan jarak antar pupil >2 SD di atas rata-rata.
Gejala sindrom noonan
Pasien datang dalam berbagai derajat:
- Hepato-splenomegali
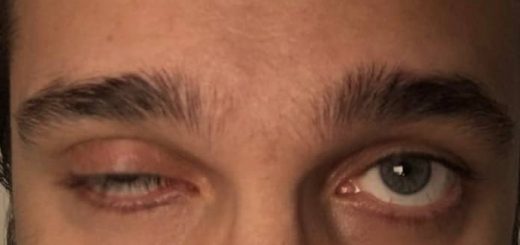
- Kelainan oftalmologis, misalnya strabismus atau ptosis
- Predisposisi rendah terhadap leukemia mielomonositik remaja
- Deformitas toraks, contohnya pectus excavatum dan pectus carinatum
- Malformasi tipe 1 Arnold-Chiari, malformasi otak kecil yang jarang terjadi
- Predisposisi langka lain untuk tumor, tanpa pemantauan khusus dianjurkan
- Kelainan genitourinari, contohnya kriptorkismus atau kelainan saluran kemih
- Penyakit jantung, biasanya stenosis katup pulmonal dan atau kardiomiopati hipertrofik
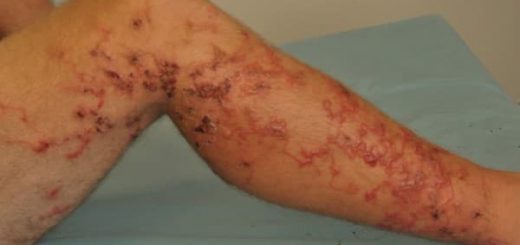
- Kelainan kulit (hiperkeratosis, rambut dan alis tipis, bintik berpigmen) dan kelainan vaskular (limfedema)
- Kelainan pendengaran THT, komplikasi infeksi telinga dan gangguan pendengaran sensorineural yang jarang terjadi
- Defisit intelektual (biasanya ringan) pada 10-20% kasus, kesulitan belajar yang tidak terkait dengan defisit intelektual juga mungkin terjadi
- Kelainan koagulasi dari gangguan sederhana pada penilaian biologis hingga kecenderungan perdarahan, tetapi sangat jarang menjadi parah
- Penundaan tinggi dan berat badan pasca kelahiran, biasanya dengan tinggi badan saat dewasa mendekati kurva -2 SD. 30% Pasien memiliki tinggi badan orang dewasa rata-rata
- Elemen morfologi minor. Wajah, misalnya hipertelorisme, mikrognatia, ptosis, telinga rendah dan miring ke belakang. Hipoplasia mandibula atau mikrognatia adalah kondisi di mana rahang bawah berukuran terlalu kecil, mikrognatia adalah gejala dari berbagai kondisi kraniofasial. Mikrognatia dapat mengganggu makanan dan pernafasan anak.

Prognosis noonan syndrome
Gambaran klinis dapat berubah yang membenarkan pemantauan khusus seumur hidup pada tingkat pusat rujukan atau kompetensi, bersama dengan dokter anak atau dokter yang merawat. Tindak lanjutnya multidisiplin dan harus dipertahankan di masa dewasa. Kemungkinan komplikasi dapat terjadi selama anestesi umum, penting untuk memberitahu ahli anestesi tentang sindrom ini sebelum menjalani operasi apapun dengan anestesi umum.
Komplikasi lain yang mungkin memerlukan perhatian khusus dan medis atau intervensi lain termasuk:
- Masalah pendarahan, penting untuk menyadari risiko ini sebelum melakukan prosedur apa pun
- Peningkatan risiko kanker, termasuk tumor jaringan lunak, kanker sel saraf dan beberapa bentuk leukemia
- Keterlambatan perkembangan, mereka mungkin memerlukan rencana khusus untuk memenuhi kebutuhan pendidikan dan perkembangan mereka
- Penumpukan cairan berlebih, kelebihan cairan yang terkumpul dari kondisi limfatik dapat menggenang di sekitar jantung dan paru-paru dan ini bisa berbahaya.
Anamnesis noonan syndrome
Diagnosis terutama bersifat klinis, konfirmasi dapat diperoleh dengan mencari mutasi pada salah satu gen yang saat ini diketahui bertanggung jawab. Pada sekitar setengah dari pasien, penyakit ini disebabkan oleh mutasi pada gen PTPN11 yang terletak pada kromosom 12. Mutasi pada gen lain dari jalur RAS (gen SOS1, RAF1, RIT1, KRAS, NRAS, BRAF, MAP2K1) telah diidentifikasi pada penderita, dalam proporsi yang bervariasi. Namun, mutasi tidak ditemukan pada sekitar 25% pasien dengan noonan syndrome.
Pada saat diagnosa, asesmen awal berupaya untuk mengetahui masalah kesehatan yang akan ditangani. Penilaian awal paling sering dikoordinasikan oleh ahli genetika klinis atau dokter anak atau dokter yang merawat. Bergantung pada komplikasi terkait, spesialis lain mungkin turun tangan. Pada semua usia, studi genetik mungkin direkomendasikan.
Penilaian tambahan berikut akan dilakukan :
- Kelainan kulit dan kelainan vaskular (limfedema) jantung (ultrasonografi + EKG dari diagnosis):
- Ultrasonografi ginjal, dari diagnosis untuk mencari kelainan saluran kemih
- Penilaian audiometri dan oftalmologi, sistematis mulai bulan ke-6, paling lambat sebelum masuk sekolah
- MRI otak dan serviks, dapat ditawarkan secara sistematis setelah 10 tahun bila terdapat disabilitas intelektual yang jelas. Dalam kasus neurological call sign pada sindrom noonan tanpa disabilitas intelektual
- Kelainan laboratorium:
- Pemeriksaan tiroid, dari usia 11 tahun
- Analisis genetik, mungkin dari diagnosis
- Jika ukuran kecil terbukti, dosis IGF1, usia tulang dan jika abnormal, tes untuk stimulasi hormon pertumbuhan
- Penilaian hematologis: CBC, trombosit dan koagulasi termasuk dosis terpisah dari faktor VIII, IX, XI dan XII (dari usia 5 tahun atau lebih awal jika operasi) dalam kasus perdarahan mudah (multipel memar), penilaian fungsi trombosit dengan pendapat hematologis diperlukan.
Pengobatan sindrom noonan
Tidak ada pengobatan kuratif khusus, tujuan terapi yang direkomendasikan adalah:
- Atur dukungan pendidikan
- Melawan malnutrisi pada bayi
- Deteksi dini dan obati komplikasi
- Pantau pertumbuhan dan kelainan jantung.
Anak-anak akan mendapat tindak lanjut secara rutin oleh dokter spesialis anak atau dokter keluarga, yang memiliki peran penting dalam:
- Memantau pertumbuhan dan status gizi
- Dukungan psikologis untuk keluarga saat diagnosis diumumkan atau jika komplikasi muncul
- Memantau perkembangan psikomotor dan membantu mengatur pendidikan atau bahkan perawatan rehabilitasi, paramedis.
Masalah medis memerlukan manajemen multidisiplin yang terpadu, beberapa spesialis dan pekerja paramedis. Direkomendasikan untuk melakukan konsultasi ringkasan tahunan (mengintegrasikan hasil dari berbagai konsultasi, status berbagai dukungan dan integrasi sekolah). Konsultasi ini harus memungkinkan dilakukannya penyesuaian tindak lanjut dan perawatan sebaik mungkin. Pusat referensi anomali perkembangan tetap menjadi teman bicara bagi para profesional dan keluarga setelah diagnosis.
Pencegahan sindrom noonan
Karena beberapa kasus noonan syndrome terjadi secara spontan, tidak ada cara yang diketahui untuk mencegahnya. Namun, jika Anda memiliki riwayat keluarga sindrom ini, bicarakan dengan dokter Anda tentang manfaat konseling genetik sebelum Anda mengandung. Noonan syndrome dapat dideteksi dengan pengujian genetik molekuler.
Referensi
- Children’s Hospital of Philadelphia : Micrognathia : https://www.chop.edu/conditions-diseases/micrognathia
- Children’s Health : Pediatric Hypertelorism : https://www.childrens.com/specialties-services/conditions/hypertelorism
- MedicalNewsToday : What’s to know about Noonan syndrome? : https://www.medicalnewstoday.com/articles/179200
- Mayo Clinic : Noonan syndrome : https://www.mayoclinic.org/diseases-conditions/noonan-syndrome/symptoms-causes/syc-20354422